More accurate transcription factor binding models
New model for the structure of transcription factor binding sites and a corresponding machine learning method developed by researchers at the University of Helsinki and Karolinska Institutet can handle large high-throughput data sets of DNA sequences.
The activity (“expression”) of genes in cells is regulated in part by so-called transcription factors (TFs). Such TFs are proteins that may bind to DNA in order to make regulatory effect on a gene associated to the binding site. To understand the regulatory mechanism in detail, it is essential to uncover for each TF the patterns or “motifs” in DNA to which the TF has strong binding affinity. Probabilistic models describing these binding affinities can be used, for example, to predict regulatory regions in a genome that are responsible for the expression of the corresponding genes.
In this research, we propose a novel modeling technique and an expectation maximization (EM) learning algorithm, implemented as software tool MODER, for discovering the modular structure of TF binding motifs. Using monomeric motifs, represented as Position Weight Matrices (PWMs), as the basic modules, the EM learning discovers both monomeric PWMs and their dimeric combinations.
The method and several new binding motifs discovered by it will be published in Nucleic Acids Research.
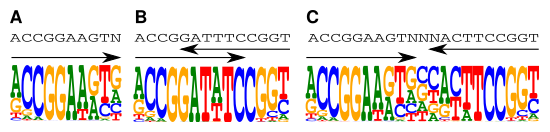
Figure: (A) A PWM model for a monomeric motif. (B, C) Two dimeric PWMs learned using the PWM (A) as the basic module.
Reference: J. Toivonen, T. Kivioja, A. Jolma, Y. Yin, J. Taipale, E. Ukkonen. Modular Discovery of Monomeric and Dimeric Transcription Factor Binding Motifs for Large Data Sets. Nucleic Acids Research, vol. 46, no. 8.
For more information:
Jarkko Toivonen, University of Helsinki, jarkko.toivonen@cs.helsinki.fi and
Esko Ukkonen, University of Helsinki, esko.ukkonen@helsinki.fi